-
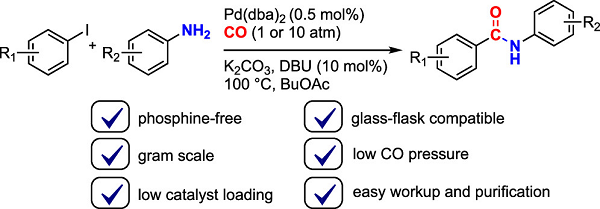
Phosphine-Free Aminocarbonylation Using Pd/DBU Catalyst: Carbonylative Coupling of Aryl Iodides and Amines
Picard, B.; Fukuyama, T.; Ryu, I.
J. Org. Chem., in press.
DOI: 10.1021/acs.joc.2c02369
An improved carbonylation method allowing amide bond formation between aryl iodides and aromatic amines is presented. In contrast to usual methods based on Pd catalysis,
this method does not require a phosphine ligand. The catalyst system simply employs bis(dibenzylideneacetone)palladium (0.5 mol %) and DBU (10 mol %). The method was
applied to the synthesis of various aromatic amides from aryl iodides and amines, and was scaled to gram order synthesis under as low as 1 atm of carbon monoxide.
-
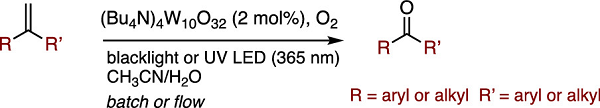
Photocatalytic Oxidative Cleavage of Alkenes by Molecular Oxygen: Reaction Scope, Mechanistic Insights, and Flow Application
Shin, Y. -L.; Wu, Y. -K.; Hyodo, M.; Ryu, I.
J. Org. Chem., in press.
DOI: 10.1021/acs.joc.2c02429
The oxidative cleavage of C═C bonds with molecular oxygen was promoted effectively by a catalytic amount of a decatungstate photocatalyst using black light irradiation (365
nm). Not only aromatic ketones but also aliphatic ketones were obtained by the photocatalytic protocol. The continuous flow reaction of α-methylstyrene using a high-power
ultraviolet light-emitting diode (365 nm) dramatically decreased the reaction time.
-
Expansion of Photocatalytic C(sp3)-H Functionalization to Allylation, Alkynylation, and Imination
Shin, Y. -L.; Huang, S. -H.; Wu, Y. -K.; Ryu, I.
Bull. Soc. Chem. Jpn., 2022, 95 , 1501-1505.
-
Bromine-Radical Mediated Bromoallylation of C-C Unsaturated Bonds. Facile Access to 1,4-, 1,5-, 1,6-, and 1,7-Dienes and Related Compounds
Sumino, S.; Ryu, I.
Synlett (Account), in press.
DOI: 10.1055/a-1998-7007
The radical bromoallylation of alkynes, allenes, and vinylidene cyclopropanes proceeds efficiently in the presence of a radical initiator to give 2-bromo-substituted 1,4-, 1,5-,
and 1,6-diene derivatives, respectively. Three-component reactions comprised of allenes, electron-deficient alkenes, and allyl bromides gives 1,7-dienes in good yields. The
bromoallylation of an arylalkene can override the β-scission of the bromine radical from β-bromoalkyl radicals to give 5-bromoalkenes. The bromoallylation of vinylcyclopropanes
is accompanied by 5-exo ring-closure to give 1-(bromomethyl)-2-vinylcyclopentane derivatives in good yields. All of the products contain a reactive vinyl-bromide moiety, which
can be readily functionalized by Pd-catalyzed cross-coupling and radical cascade reactions.
-
Pd-Mediated Light-Controlled Living Radical Polymerization of Methyl Acrylate
Sumino, S.; Ryu, I.
Bull. Soc. Chem. Jpn. (BCSJ Award Article), 2022, 95, 1532-1536.
DOI: 10.1246/bcsj.20220230
The radicalpolymerization ofmethylacrylatein the pres-ence ofapalladium dimer complex and ethyliodoacetate as aninitiator under photoirradiation conditionsled to the
formationofpoly(methylacrylate) having good Ð values. The polymer-ization could be controlled by switching thelight on and off,exhibitingliving characteristics. We propose that
suchα-Pdpolyesters couldfunction as dormant species, which wouldallow a pairoftransientα-keto radicals and persistent Pd radi-cals to be generated upon photoirradiation.
%202022%2095%201532-1536%20small%20px.png)
-
Cross-coupling reactions catalyzed by RuHCl(CO)(PPh3)3
Fukuyama, T.; Picard, B.; Ryu, I.
Catalysis, Science & Technology. (mini-review), in press.
DOI: 10.1039/D2CY01953A
Along with its periodic table neighbors, ruthenium has been extensively explored as a catalyst and found to possess a rich chemistry. Specifically, ruthenium hydrides have
caught the attention of our group for a long time, thanks to the various reactions they can catalyze. In this minireview, we will summarize our efforts made over the last two
decades toward the development of new catalytic methods for the formation of C–C bonds. In particular, the versatility of RuHCl(CO)(PPh3)3 (or RuH for short) as a commercially
available catalyst will be illustrated by eight different transformations. The elementary steps involved in the catalysis will be highlighted to demonstrate how our mechanistic
understanding in combination with the diverse catalytic capabilities of RuH can be used to design desired catalytic cycles and develop new crosscoupling reactions with high atom-
economy.
-
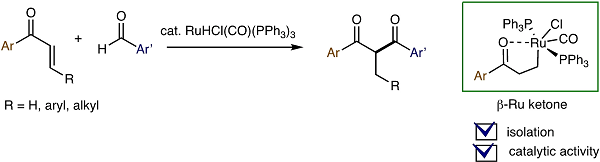
RuH-Catalyzed Synthesis of 1,3-Diaryl Ketones via Cross-Coupling of Aromatic Enones with Aromatic Aldehydes and Mechanistic Insights
Kuwahara, T.; Fukuyama, T.; Picard, B.; Ryu, I.
Adv. Synth. Catal. 2022, 364 , 3725-3729.
DOI: 10.1002/adsc.202200765
RuHCl(CO)(PPh3)3-catalyzed cross-coupling of alkenyl ketones with aldehydes has been extended to the synthesis of 1,3-diaryl 1,3-diketones. The reaction has been
successfully applied to the synthesis of PPT (propyl pyrazole triol), a selective estrogen receptor modulator compound. A study of the stoichiometric reaction of phenyl vinyl
ketone with RuHCl(CO)(PPh3)3 has revealed that the β-ruthenium ketone PhCOCH2CH2RuCl(CO)(PPh3)2, a five-membered ruthenacycle is formed with liberation of one
molecule of PPh3. The observed catalytic activity of this ruthenacycle in the presence of PPh3 suggests that it serves as a resting state, in equilibrium with an oxa-π-allyl
ruthenium species, which is the key coupling partner with aldehyde substrates.
-
New directions in radical carbonylation chemistry: combination with electron catalysis, photocatalysis and ring-opening
Kawamoto, T.; Fukuyama, T.; Picard, B.; Ryu, I.
Chem. Commun. 2022, 58 , 7608-7617.
DOI: 10.1039/d2cc02700c
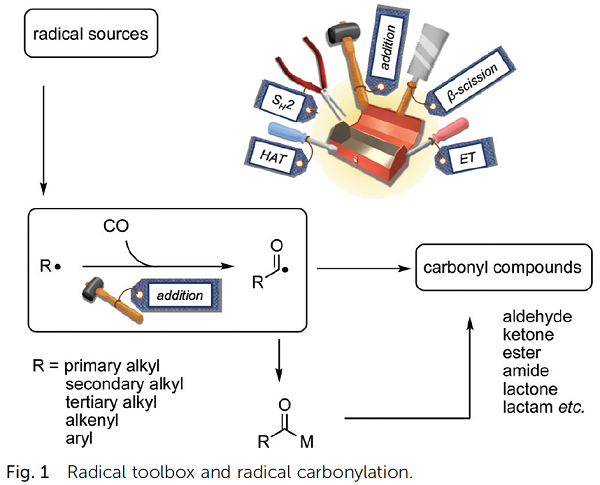
Radical carbonylation offers potent methods for introducing carbon monoxide into organic molecules. This feature article focuses on our current efforts to develop new
strategies for radical carbonylation, which include electron-transfer carbonylation, site-selective C(sp3)–H carbonylation by a photocatalyst and ring-opening carbonylation.
-
Improved efficiency of photo-induced synthetic reactions enabled by advanced photo flow technologies
Fukuyama, T.; Kasakado, T.; Hyodo, M.; Ryu, I.
Photochem. Photobiol. Sci. 2022, 21 , 761-775.
DOI: 10.1007/s43630-021-00151-6
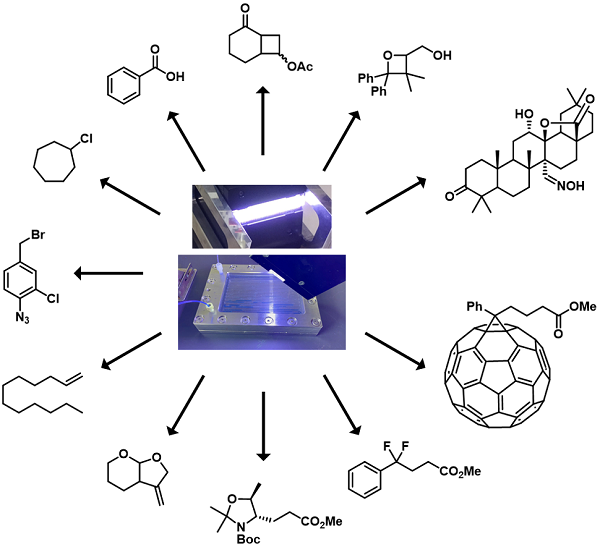
In this article, we discuss how effective photo-induced organic reactions became when applied evolving photo flow technologies through our experiences over these two last
decades. We started with the flow update of traditional [2 + 2] cycloaddition using Mikroglas Dwell device as a flow reactor and a compact light source, such as blacklight, instead
of a high-pressure mercury lamp. Then we examined Barton nitrite reaction using a photo flow reactor consisting of stainless-steel channels and a quartz glass top provided by
DNS. Again the use of blacklight was successful. However, the energy profile of these reactions was improved further by the use of LED lights. We used a photo-flow set-up,
consisting of stainless steel engraved microchannels covered by a quartz top (MiChS L-1) and a sodium lamp, for the isomerization of a fulleroid to PCBM. Photo-redox-catalyzed
alkene alkylation proceeded within a shortened reaction time when the same photo flow reactor and white LED were used instead of a batch reactor. Photo-induced reductive 5-
exo-dig radical cyclization and reduction of alkenyl halides proceeded smoothly, thanks to the combination of a photo flow reactor and low-pressure Hg lamp. We also applied flow
technologies for photo-bromination and chlorination of C–H bonds. Photocatalytic oxidation of benzyl alcohol by molecular oxygen became quick when high-power LED irradiation
was employed.
-
High-speed C-H chlorination of ethylene carbonate using a new photoflow setup
Kasakado, T.; Fukuyama, T.; Nakagawa, T.; Taguchi, S.; Ryu, I.
Beilstein J. Org. Chem. 2022, 18 , 152-158.
DOI: 10.3762/bjoc.18.16
We report the high-speed C–H chlorination of ethylene carbonate, which gives chloroethylene carbonate, a precursor to vinylene carbonate. A novel photoflow setup designed
for a gas–liquid biphasic reaction turned out to be useful for the direct use of chlorine gas. The setup employed sloped channels so as to make the liquid phase thinner, ensuring a
high surface-to-volume ratio. When ethylene carbonate was introduced to the reactor, the residence time was measured to be 15 or 30 s, depending on the slope of the reactor
set at 15 or 5°, respectively. Such short time of exposition sufficed the photo C–H chlorination. The partial irradiation of the flow channels also sufficed for the C–H chlorination,
which is consistent with the requirement of photoirradiation for the purpose of radical initiation. Near-complete selectivity for single chlorination required the low conversion of
ethylene carbonate such as 9%, which was controlled by limited introduction of chlorine gas. At a higher conversion of ethylene carbonate such as 61%, the selectivity for
monochlorinated ethylene carbonate over dichlorinated ethylene carbonate was 86%. We found that the substrate contamination with water negatively influenced the
performance of the C–H chlorination.

-
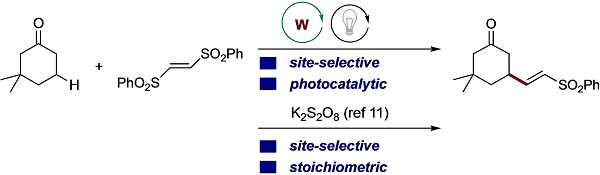
Site-Selective C(sp3)-H Alkenylation Using Decatungstate Anion as Photocatalyst
Wang, Y. -T.; Shih, Y. -L.; Wu, Y. -K.; Ryu, I.
Adv. Synth. Catal. 2022, 364 , 1039-1043.
DOI: 10.1002/adsc.202101374
Site-selective C(sp3)-H alkenylation was achieved under photo-irradiation in the presence of a catalytic amount of decatungstate anion, W10O324−. In this reaction, the radical
addition/β-scission sequence is successfully combined with photocatalytic C(sp3)-H functionalization. The reaction using weaker C−H bonds such as those in THF revealed that
the benzenesulfonyl radical itself underwent HAT directly from the C−H bond, and a decatungstate anion participated in a chain-repairing step.