The Projects ongoing within our Laboratory focus on inventing new radical reactions and catalysis, which could be potentially valuable in organic synthesis. Unlike ionic
reactions, free-radical reactions can override tedious protection/deprotection issues. Radical reactions require only a small amount of initiator, which may be replaced by simple
heating and photoirradiation. These features fit the late-stage functionalization, which is essential for the straightforward synthesis of biologically active compounds and functional
materials.
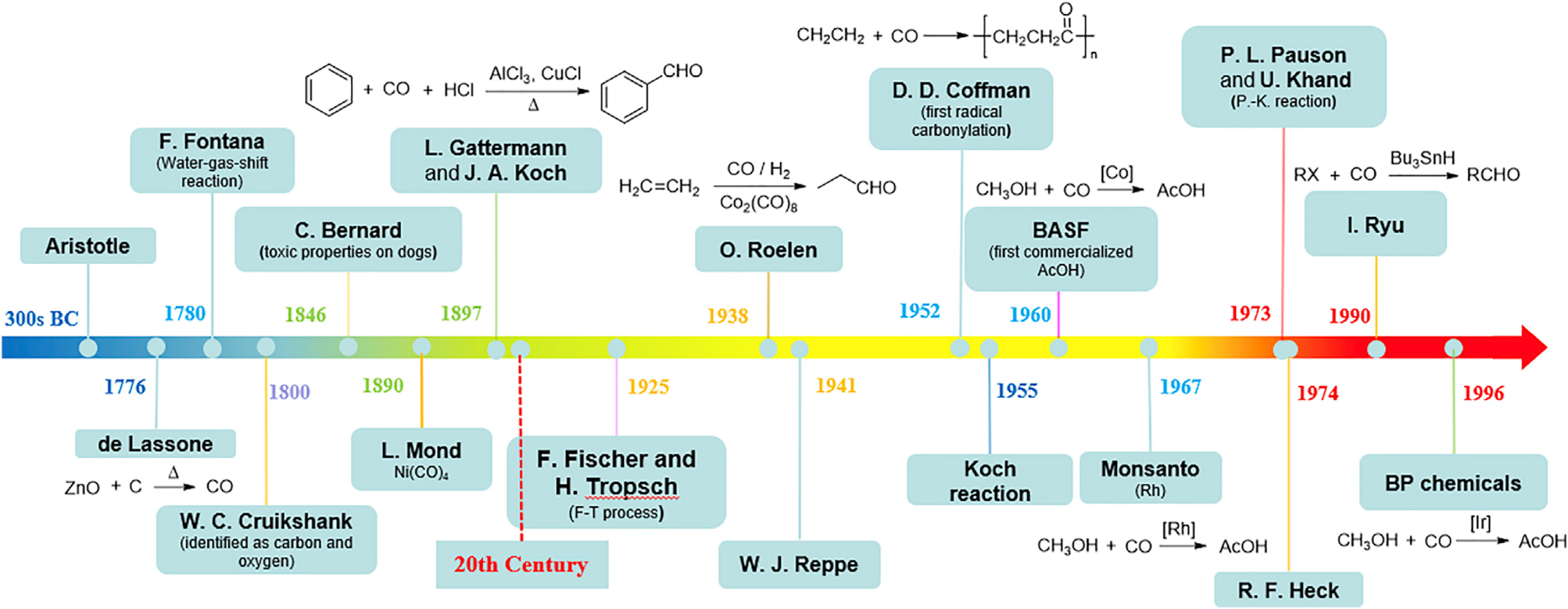
History of CO Chemistry (Taken from Xiao-Feng Wu’s review: Chem. 2019, 5, 526–552.)
CO is a cheap feedstock produced from coal and steam and the development of the methods to convert CO2 to more reactive CO is actively being pursued. Powerful
methodologies for the incorporation of CO into organic molecules (carbonylation) have been among long-standing themes., as seen in the History Table taken from Xiao-Feng
Wu's recent review. Some of the carbonylation processes met with industrial success, which include acetic acid synthesis from MeOH and CO by Rh and Ir catalyst
(Monsanto/Cativa process) and hydroformylation of alkenes by CO and molecular hydrogen (oxo process). In this important area, we strongly focused on radical carbonylation,
which had been neglected for many years plagued by the low yield reactions reported in the earlier stage.
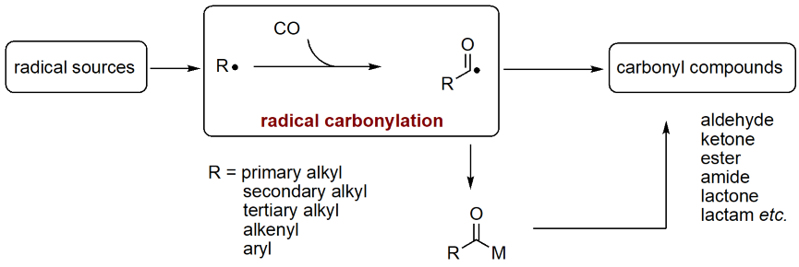
The Ryu group has pioneered the field of radical carbonylation, where acyl radicals as key intermediates. Radical carbonylation accommodates cascade reactions to couple
with alkenes, alkynes, and nitrogen-containing compounds in a single operation.
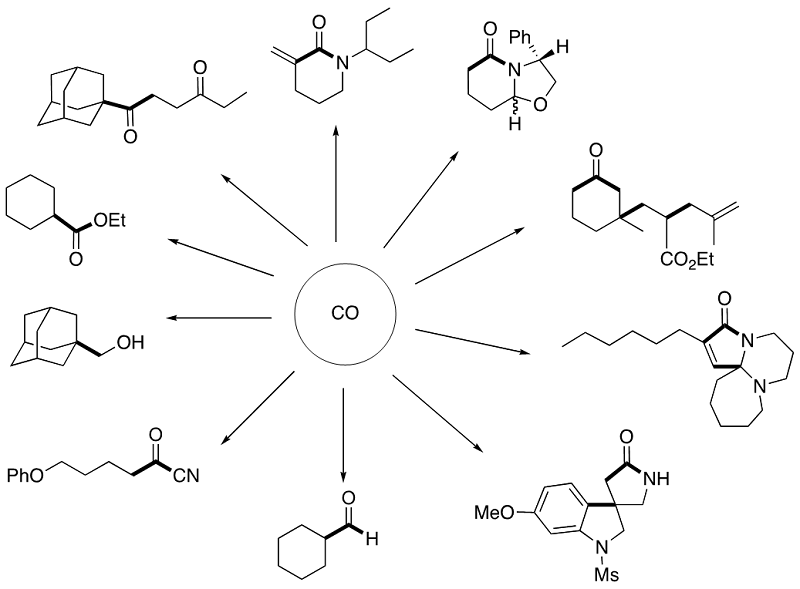
Thus far, we have successfully developed a variety of methods to incorporate CO into organic molecules as a carbonyl function. The concept and examples are summarized in
the following two Schemes.
For reviews on radical carbonylation, see:
(a) Ryu, I.; Sonoda, N. Angew. Chem. Int. Ed. 1996, 35, 1050.
(b) Ryu, I.; Sonoda, N.; Curran, D. P.; Chem. Rev. 1996, 96, 177.
(c) Ryu, I. Chem. Soc. Rev. 2001, 30, 16.
(d) Ryu, I. Chem. Rec. 2002, 2, 249.
(e) Ryu, I.; Uenoyama, Y.; Matsubara, H. Bull. Chem. Soc. Jpn. 2006, 79, 1476.
(f) Schiesser, C. H.; Wille, U.; Matsubara, H.; Ryu, I. Acc. Chem. Res. 2007, 40, 303.
(g) Sumino, S.; Fusano, A.; Fukuyama, T.; Ryu, I. Acc. Chem. Res. 2014, 47, 1563.
(h) Fusano, A.; Ryu, I. In SOS: Multicomponent Reactions 2; Muller, T. J. J. Eds.; Georg Thieme Verlag KG, Germany, 2014.
(i) Matsubara, H.; Kawamoto, T.; Fukuyama, T.; Ryu, I. Acc. Chem. Res. 2018, 51, 2023.
(j) T. Kawamoto, T. Fukuayma, B. Picard, I. Ryu, Chem. Comm. (feature article) submitted.
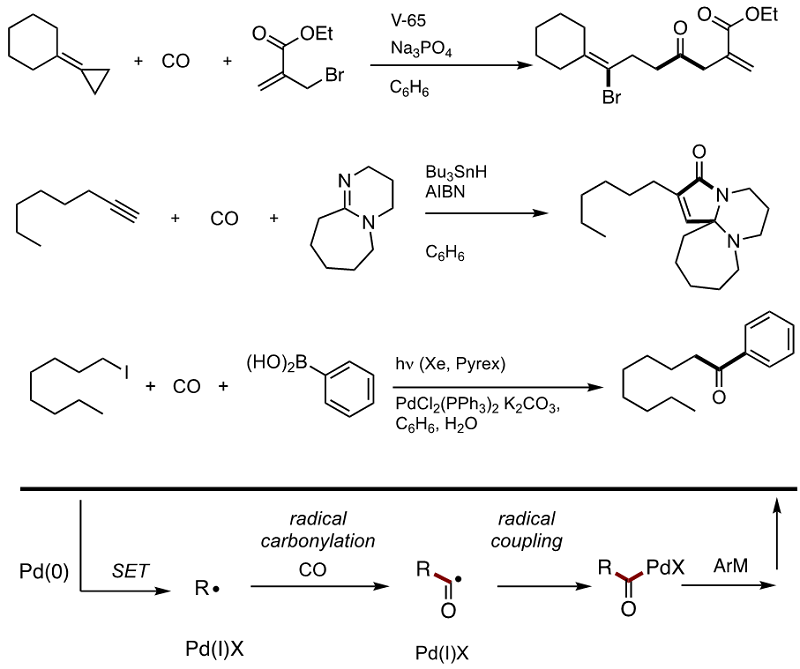
Some new progresses are also highlighted. The first reaction is carbonylative bromoallylation, in which the radical arising from bromine radical addition leads to the key alkyl
radical via beta-scission. The second is a formal [2+2+1] cycloaddition, in which tributyltin radical mediates the multi-step reactions as a catalyst. The third example is a
radical/Pd hybrid carbonylation leading to alkyl aryl ketones, in which radical persistent effects by PdX radical works as the key. Now we are working on
electron transfer carbonylation,
for which neither radical mediator nor metal catalyst is necessary (the fourth example). Based on the general concept of the
electron as a catalyst,
advocated by Studer, Curran, and Shirakawa, we are keen to develop novel radical cascade reactions.
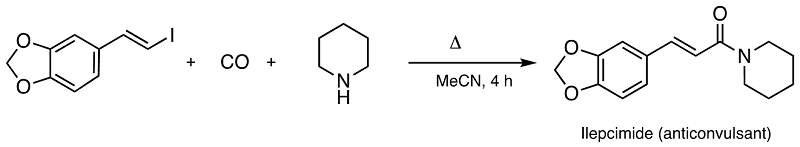
(a) T. Kippo, K. Hamaoka, I. Ryu, J. Am. Chem. Soc. 2013, 135, 632-635.
(b) T. Fukuyama, N. Nakashima, T. Okada and I. Ryu, J. Am. Chem. Soc. 2013, 135, 1006-1008.
(c) S. Sumino, T. Ui and I. Ryu, Org. Lett. 2013, 15, 3142-3145.
(d) T. Kawamoto, A. Sato and I. Ryu, Chem. Eur. J. 2015, 21, 14764–14767.
(e) B. Picard, T. Fukuyama, T. Bando, M. Hyodo and I. Ryu, Org. Lett. 2021, 23, 9505–9509.
Catalysis
continuously stays as important challenges in chemical science no matter what catalytic species are chosen
.
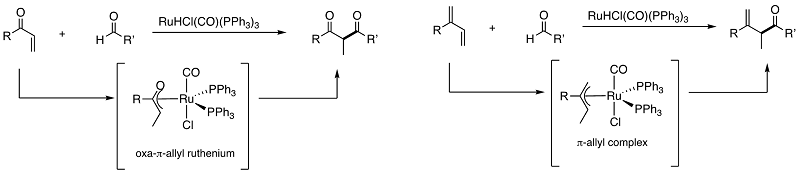
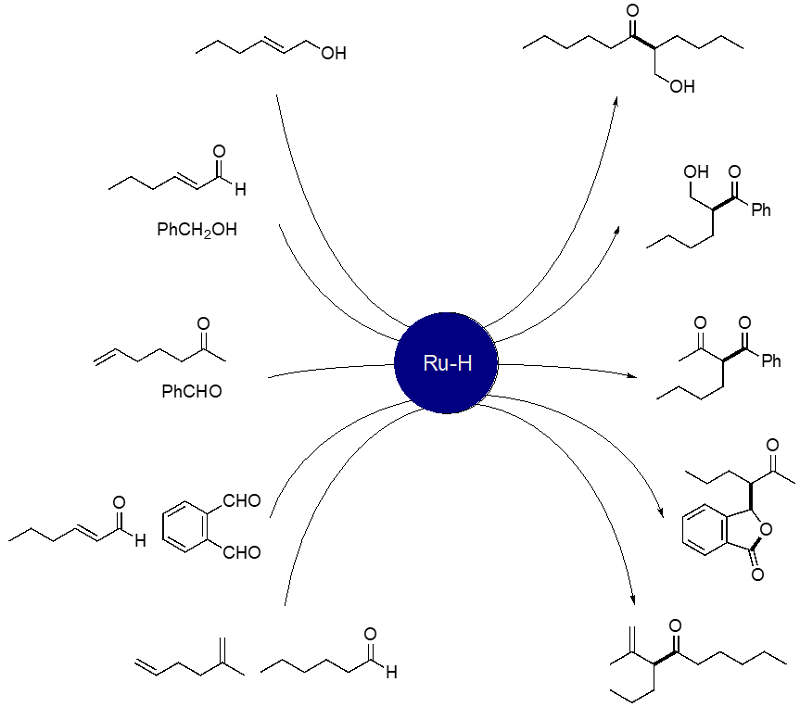
We have found that ruthenium hydride complexes serve as an efficient catalyst for atom-economical cross-coupling reactions. The following examples show the catalytic
synthesis of β-diketones and β,γ-unsaturated ketones. Since RuH causes alkene isomerization, the combined isomerization/cross-coupling sequence is possible. The other
important feature of RuH catalysis is to catalyze facile transfer hydrogenation from alcohols. The following Scheme summarizes our achievements in this area. We are
preparing a mini-review on
this topic shortly.
(a) Fukuyama, T.; Doi, T.; Minamino, S.; Omura, S.; Ryu, I. Angew. Chem. Int. Ed. 2007, 46, 5559. Cited: 75
(b) Omura, S.; Fukuyama, T.; Horiguchi, J.; Murakami, Y.; Ryu, I. J. Am. Chem. Soc. 2008, 130, 14094. Cited: 151
(c) Doi, T.; Fukuyama, T.; Minamino, S.; Husson, G.; Ryu, I. Chem. Commun. 2006, 1875.
(d) Doi, T.; Fukuyama, T.; Horiguchi, J.; Okamura, T.; Ryu, I. Synlett 2006, 721.
(e) Doi, T.; Fukuyama, T.; Minamino, S.; Ryu, I. Synlett 2006, 3013.
(f) Omura, S.; Fukuyama, T.; Murakami, Y.; Okamoto, H. Ryu, I. Chem. Commun. 2009, 6741.
(g) Denichoux, A.; Fukuyama, T.; Doi, T.; Horiguchi, J.; Ryu, I. Org. Lett. 2010, 12, 1.
(h) Fukuyama, T.; Okamoto, H.; Ryu, I. Chem. Lett. 2011, 40, 1453.
(i) Kuwahara, T.; Fukuyama, T.; Ryu, I. Org. Lett. 2012, 14, 4703. Cited: 92
(j) Kuwahara, T.; Fukuyama, T.; Ryu, I. Chem. Lett. 2013, 42, 1163.
(k) Kuwahara, T.; Fukuyama, T.; Ryu, I. RSC Adv. 2013, 3, 13702.
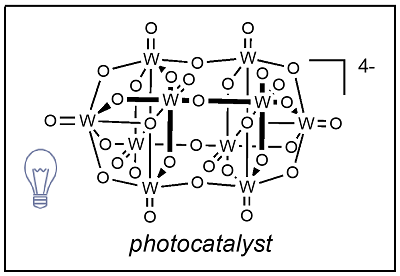
We are also very keen to the use of photocatalyst (PC).
Site-selective C(sp3)-H functionalization
is a holy grail in modern organic synthesis and we believe an excellent potential of radical substitution (SH2) reactions. The Barton nitrite reaction, enabling remote
functionalization of saturated alcohols has a long history. The driving force is to form strong O-H bond (>105 kcal/mol) at the cost of weaker C-H bond cleavage
(up to 100 kcal/mol). Intramolecularity by entropy factor is a key for site-selective delta-C-H cleavage.
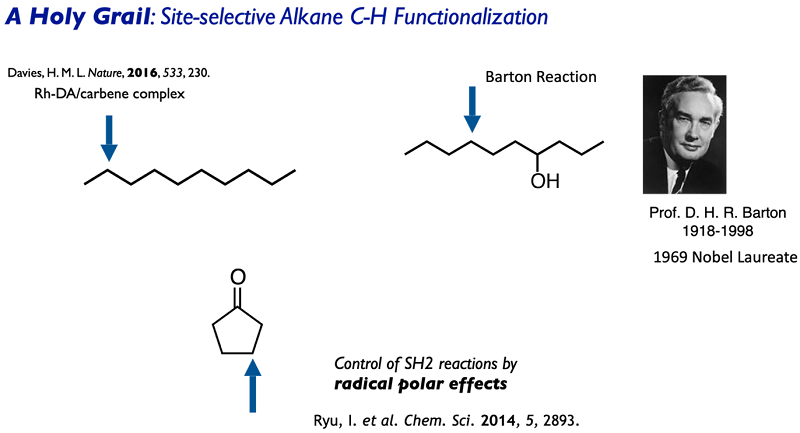
In contrast, intermolecular site-selective C-H functionalization has a higher hurdle. A breakthrough was brought by the study in our Lab. In the case of cyclopentanone, we
found that weaker beta-C-H bond is selectively cleaved and we propose radical polar effects in the HAT transition state between C-H bonds and oxygen-centered radicals. The
strategy based on polar effects and stereo effects proved to be useful to achieve site-selective C-H functionalization and many other groups applied the strategy now.
Decatungtate anion is such a photocatalyst (PC) and we had many fruitful collaborations with the Pavia group.
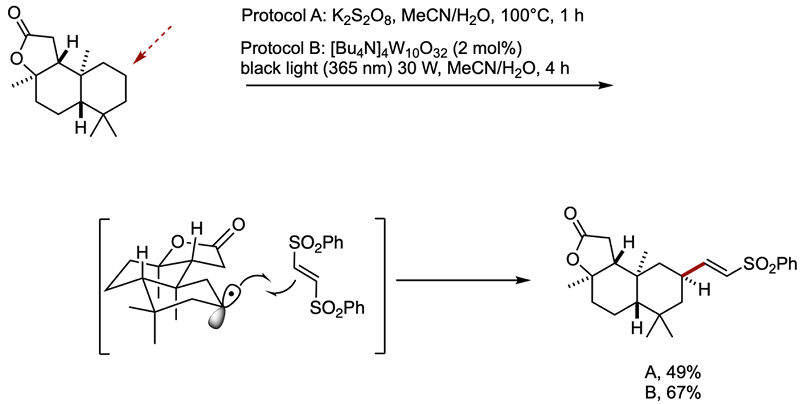
The C-H alkenylation was examined by two protocols, stoichiometric and catalytic. The following remarkable case features the selection of one out of 26 CH bonds of (3aR)
(+)- sclareolide.
(a) For a key review, see: D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa, I. Ryu, ACS Catal. 2018, 8, 701–713. cited: 191
(b) T. Fukuyama, T. Nishikawa, I. Ryu, Eur. J. Org. Chem. 2019, 2020, 1424–1428.
(c) T. Fukuyama, K. Yamada, T. Nishikawa, D. Ravelli, M. Fagnoni, I. Ryu, Chem. Lett. 2018, 47, 207–209.
(d) K. Yamada, T. Fukuyama, S. Fujii, D. Ravelli, M. Fagnoni, I. Ryu, Chem. Eur. J. 2017, 23, 8615–8618.
(e) T. Fukuyama, T. Nishikawa, K. Yamada, D. Ravelli, M. Fagnoni, I. Ryu, Org. Lett. 2017, 19, 6436–6439.
(f) M. Ueda, K. Kamikawa, T. Fukuyama, Y. -T. Wang, Y. -K. Wu, I. Ryu, Angew. Chem. Int. Ed. 2021, 60, 3545–3550.
(g) Y. -T. Wang, Y. -L. Shih, Y.-K. Wu, I. Ryu, Adv. Synth. Catal. 2022, 364, 1039-1043.
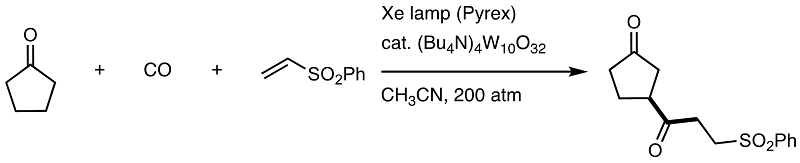
PC-Controlled C-H carbonylation
is within our recent interest. In collaboration with the Pavia group, we already reported on C-H carbonylation using decatungstate anion as PC catalyst in 2011, which has been
updated, having the site-selective case leading to unsymmetrical ketones. For C-H carbonylation DIAD can be used as an acyl radical trap, giving amides and aliphatic nitriles
also accommodate the site-selective C-H carbonylation.

(a) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni, A. Albini, Angew. Chem. Int. Ed. 2011, 50, 1869–1872.
(b) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro, M. Fagnoni, Org. Lett. 2013, 15, 2554–2557.
(c) M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli, M. Fagnoni, Chem. Sci. 2014, 5, 2893–2898.