English
Research
We are working on the development of catalysts and functional molecular systems that support sustainable life, health, and beauty for people under the motto "Have fun, be spontaneous, and have a dream" and with a focus on human resources development, advancement of science, and creating shared value.
The development of innovative functional molecular systems requires an accurate understanding of unique phenomena and a discussion of the underlying principles and mechanisms, e.g., deep considerations grounded in coordination chemistry and organic chemistry. In our laboratory, we are developing highly creative research based on the background of fundamental fields, making the best use of technology, knowledge and experience in coordination chemistry, organometallic chemistry, organic chemistry, biology and materials chemistry.
Development of Environmentally-Benign Catalytic Systems
The precious metals that have been used as catalysts in the past are in danger of depletion, so the development of efficient catalytic systems that do not use rare metals is an important issue for the future. Aiming for a sustainable global environment, we are developing environmentally-benign catalytic systems and biomass conversion technologies using commercially available inexpensive vanadium and iron catalysts with high clarke counts.
Early transition metal vanadium, considered one of the essential elements in biological systems, is widely distributed in the biological environment, and its compounds are involved in the control of various reactions in the biological system. Vanadium is also a metal with a high clarke count and is commercially available. Because of its high Lewis acidity, various redox states, and oxygen affinity, vanadium is expected to expand into novel organic reactions and especially redox reactions. Iron is the fourth most abundant in the earth's crust and is very cheap. Because of its high Lewis acidity and redox properties, it is expected to be developed as an alternative metal catalyst to rare metals. From these points of view, we are developing environmentally-benign catalytic systems based on Lewis acidity, redox properties, and oxygen affinity of vanadium and iron centers.
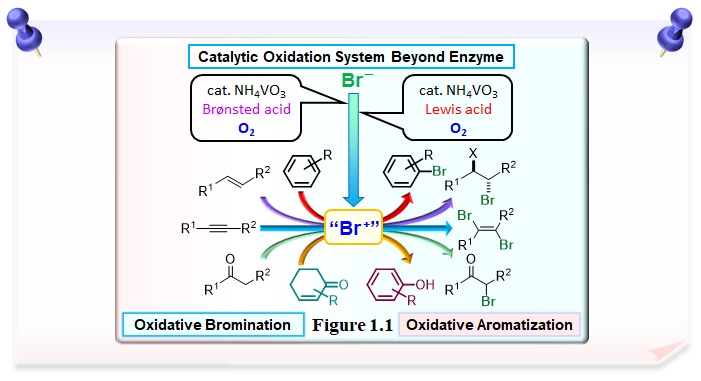
By incorporating the redox properties of vanadium into the catalytic cycle, we have successfully developed environmentally friendly oxidative bromination and oxidative aromatization catalysts beyond enzymes(Figure 1.1)。That is, the catalytic conversion of a bromide ion to a brominium cation-like species using molecular oxygen. In addition, the combination of halide ions and Lewis acid allows for catalytic chlorination and iodization.
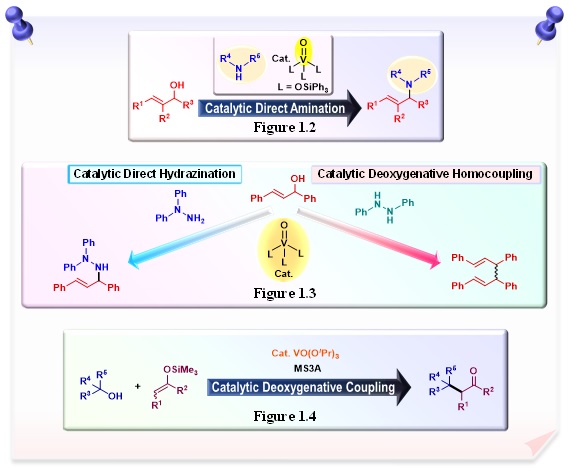
By using the Lewis acidity and oxophilicity of oxovanadium(V) complexes, we are developing a catalytic system for the direct amination of allyl alcohols (Figure 1.2). The only by-product is water, and this catalytic system is a clean catalyst system that reduces the generation of unwanted waste.
Utilizing the Lewis acidity, redox properties and oxygen affinity of vanadium catalysts, we have also successfully developed a catalytic system for the deoxygenative homocoupling reaction of alcohols without the need for a stoichiometric amount of metal reductants (Figure 1.3). Deoxygenative coupling reactions are noteworthy from the point of view of biomass utilization and are expected to contribute to the development of biomass conversion technologies.
Furthermore, by taking advantage of the Lewis acidity, redox properties, and oxophilicity of the vanadium catalyst, which has a high Clark number and are commercially available, we have developed a catalytic system for the deoxygenative coupling reaction of allyl alcohols with trimethylsilyl enol ethers to afford g,d -unsaturated carbonyl compounds in one-step (Figure 1.4). The g,d -unsaturated carbonyl compounds are important building blocks in natural product synthesis. This catalytic system can be applied to the deoxygenative coupling reaction of benzyl alcohols with trimethylsilyl enol ethers, providing the corresponding carbonyl compounds.
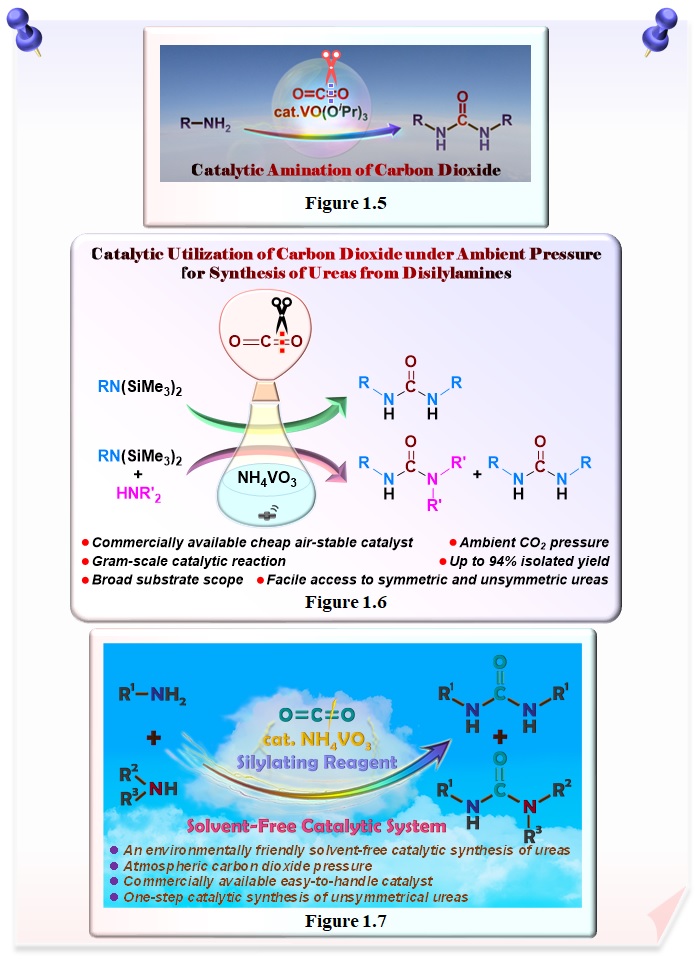
Carbon dioxide, which is a non-toxic, abundant, and green carbon resource, is regarded as a reliable C1 building block in organic synthesis. The development of methodologies for the catalytic transformation of carbon dioxide into valuable compounds is of fundamental importance for the future sustainable society. We have successfully developed a catalytic system for urea synthesis based on the catalytic activation of carbon dioxide using in situ generated imidovanadium(V) compounds from amines and commercially available oxovanadium(V) compounds (Figure 1.5).
A commercially available easy-to-handle NH4VO3 has been successfully used to develop a catalytic system for the synthesis of ureas from disilylamines by the catalytic activation of carbon dioxide as a C1 building block under ambient pressure (Figure 1.6). This is the first example of the catalytic synthesis of ureas from disilylamine and carbon dioxide under ambient pressure. Furthermore, by exploiting the difference in reactivity between disilylamines and amines, we have succeeded in catalytic synthesis of unsymmetrical ureas.
An environment-friendly, solvent-free catalytic synthesis of ureas in one-step from amines and carbon dioxide under atmospheric pressure was successfully achieved by using a commercially available air-stable NH4VO3 catalyst in the presence of BSA as a silylating reagent and also a solvent (Figure 1.7). This catalytic system was applicable to one-step preferential synthesis of unsymmetrical ureas.
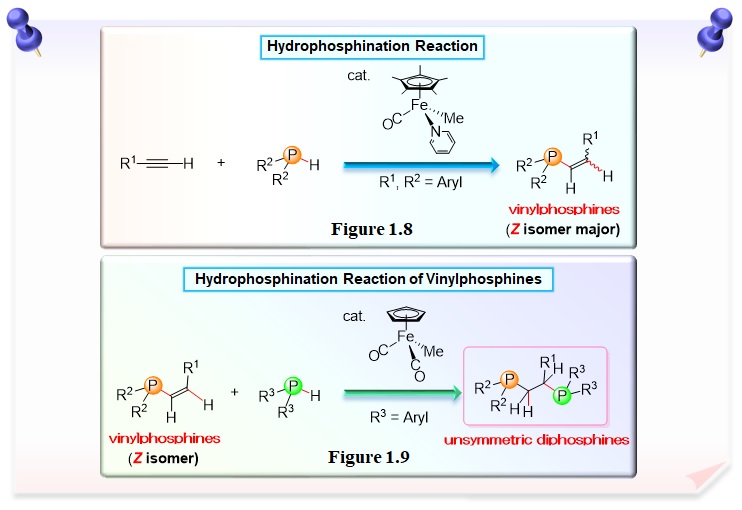
Trivalent phosphorus compounds usually act as a catalyst poison because of lone pair. However, we found that regioselective hydrophosphination of terminal arylalkyne with secondary phosphine was achieved in the presence of an iron catalyst (Figures 1.8 and 1.9). This result has attracted worldwide attention as a simple synthetic method of useful bidentate phosphine compounds and follow-up researches using other transition-metal catalyst are actively conducted.
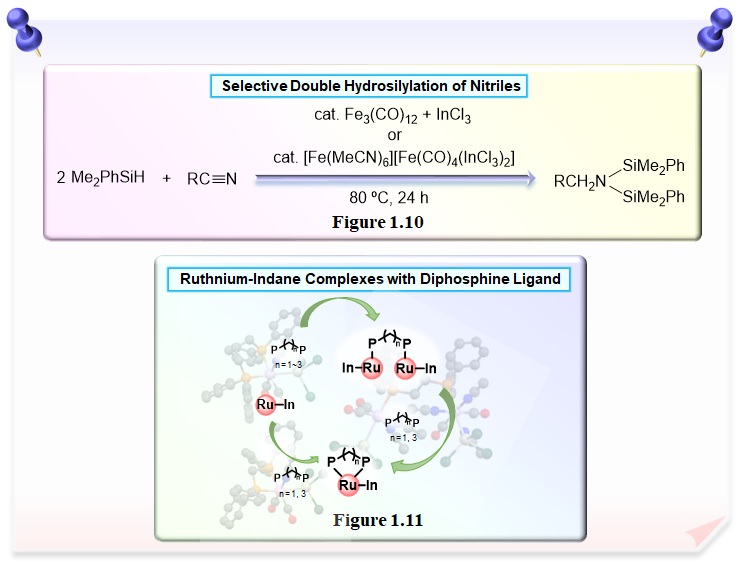
The first example of selective double hydrosilylation of the C≡N bond of organonitriles was achieved in the presence of a catalytic amount of triirondodecacarbonyl [Fe3(CO)12] and indium trichloride (InCl3) and disilylamines were obtained. This is a novel report of the combination of a transition-metal complex and an indium source in organic synthesis (Figure 1.10). We found reaction of ruthnium-indane complex with diphosphine ligand afforded the more stable chelate complex via dinuclear ruthenium-indane complex linked by diphosphine ligand (Figure 1.11). Furthermore, these complexes act as a catalyst for the double-hydrosilylation of organonitrile.
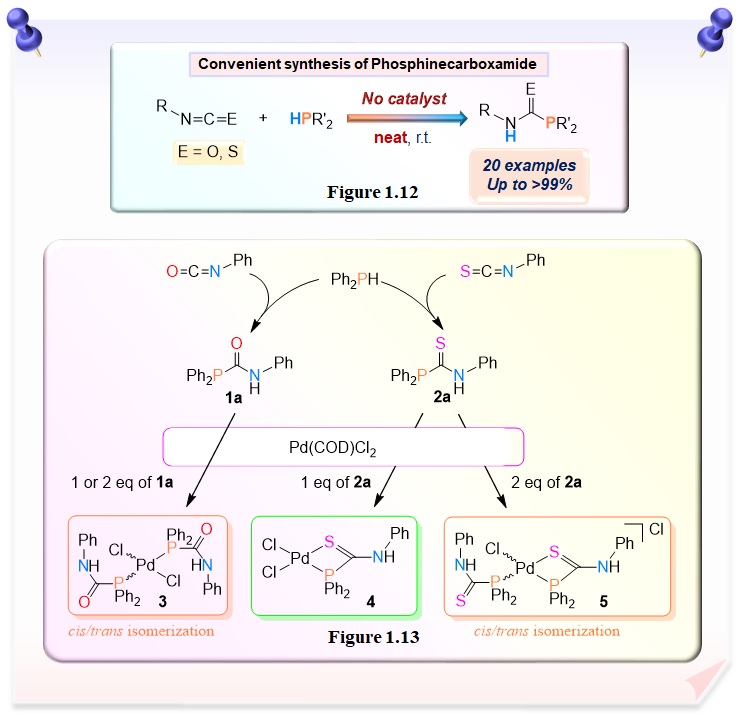
Recently, we achieved that convenient synthesis of phosphinecarboxamide by hydrophosphination of secondary phosphine with isocyanate was achieved under mild conditions without catalyst and solvent. This system shows shorter reaction time, high yield, and good functional group tolerance. Therefore, we considered that our reaction is also useful from the viewpoint of green chemistry and sustainability (Figure 1.12). We also examined their complexation with palladium(II) complex (Figure 1.13).
Functional Design of Hybrid Molecular Systems Created by Biomolecules
We are pioneering an unexplored field of research in bioorganometallic chemistry that combines biology and organometallic chemistry. By applying nature-created nanotechnology, we are establishing functionally emergent molecular alignment methods and creating hybrid molecular systems with novel functional properties.
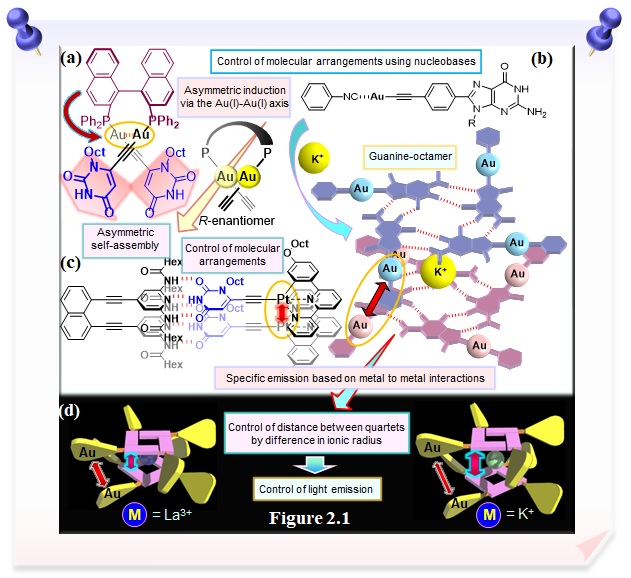
The self-assembly program of nucleobases is also being used to control molecular arrangements. We are the first to successfully induce chirality in the Au(I)-Au(I) axis by introducing the chiral (R)-BINAP as a bridging diphosphine ligand with axial chirality in a dinuclear organogold(I) complex with the uracil moieties, allowing the formation of a helical assembly based on hydrogen bonds between the uracil moieties (Figure 2.1a). In the guanosine-based Au(I) isonitrile complex, chirality organization of the guanine moiety leads to the formation of G-octamer, allowing for specific emission based on Au(I)-Au(I) interaction (Figures 2.1b and 2.1d), revealing the utility of nucleobase self-assembly in constructing specific reaction fields. We have also succeeded in controlling the spatial arrangement of the bioorganometallic uracil-based platinum(II) compounds using complementary base pairing, which allows functional control of specific emission based on metal-to-metal interactions (Figure 2.1c). Furthermore, we have shown that nucleobase-conjugated complexes exhibit high cytotoxicity.
Construction of Chirality Organized Supramolecular Systems
The ability to control the directionality, specificity, reversibility, tuneability, and dynamic nature of hydrogen bonds would allow for the development of functional hybrid molecular systems with both flexibility and dynamic properties, as seen in biological systems. By utilizing the chirality organization properties of amino acids and nucleobases, which are biomolecules that can be arranged and combined in various ways, we are developing unique chirality-organized hybrid systems that are conceptually different from the conventional asymmetric fields, as well as specific reaction fields.
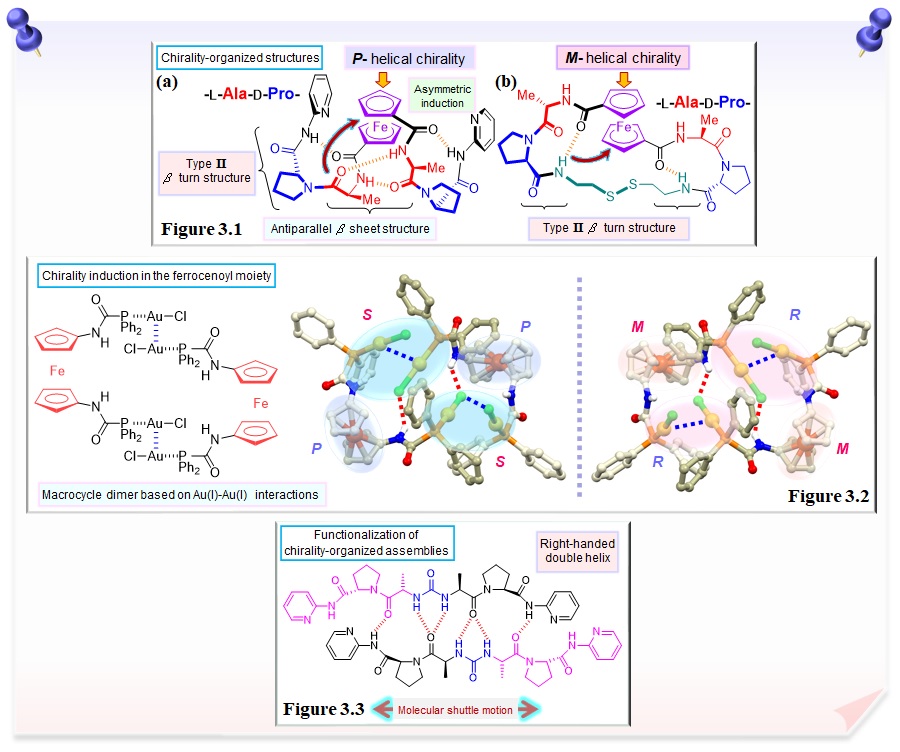
We have succeeded in controlling the formation of protein secondary structures such as β sheet and β turn structures by simply introducing a dipeptide chain, the smallest unit of a peptide, into ferrocene with a reversible redox couple and two rotatory coplanar cyclopentadienyl rings as a molecular scaffold with dynamic functions (Figure 3.1a). The helical chirality of the ferrocenoyl moiety is found to be induced by chirality organization based on intramolecular hydrogen bonds in the dipeptide chain. We have also succeeded in controlling the formation of protein secondary structures and chirality-organized structures by controlling the sequence and combination of amino acids and the complexation with metal salts. By structurally regulating the dipeptide chains into a cyclic structure with disulfide bonds, controlling helical chirality in the ferrocene-dipeptide bioorganometallic conjugates are achieved without altering the chirality of the amino acids (Figure 3.1b). The conformationally regulated ferrocene-peptide conjugate provides the chirality-organized binding site for size-selective and chiral recognition of dicarboxylic acids through multipoint hydrogen bonds. These studies are noted as pioneering research in the field of bioorganometallic chemistry.
We have succeeded in synthesizing a macrocyclic gold(I) complex, which is driven by Au(I)-Au(I) interactions, by the reaction of 1,1'-bis(diphenylphosphinecarboxamidyl)ferrocene with chloro(tetrahydrothiophene)gold(I), [AuCl(tht)] (Figure 3.2). In addition, the use of chiral amino acid derivatives as guest molecules allows for the chirality induction in the ferrocenoyl moiety.
We are also working on the functionalization of chirality-organized assemblies using urea derivatives as a molecular scaffold, which are capable of multi-point hydrogen bonding. Urea-dipeptide conjugates form a right-handed duplex by chirality organization based on intermolecular hydrogen bonds and that molecular shuttle motion based on the recombination of hydrogen bonds is possible (Figure 3.3).
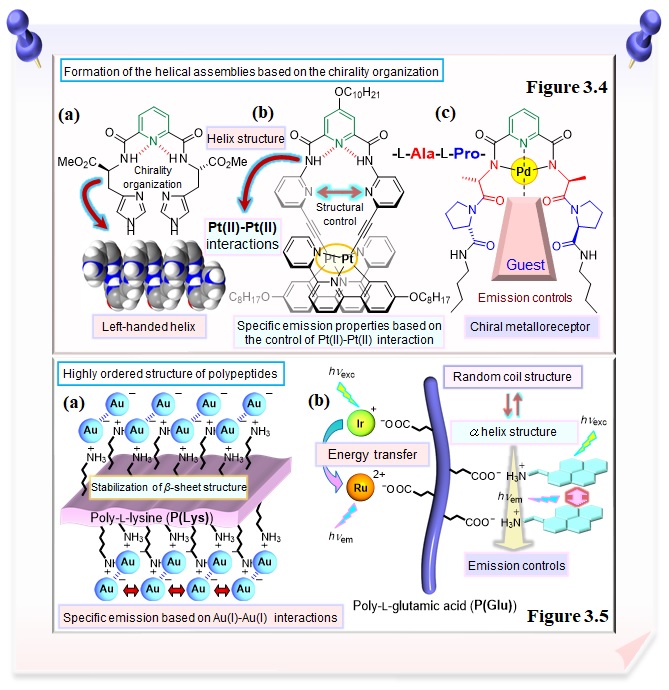
The introduction of amino acids into the pyridine molecular scaffold leads to the formation of the helical assemblies based on the chirality organization of amino acids. The difference in the chirality of the amino acids allows directional control of the helical structure (left-handed/right-handed) (Figure 3.4a). Also, by controlling the structure of the helical structure, we have successfully controlled the specific emission properties based on the control of Pt(II)-Pt(II) interaction (Figure 3.4b). The chirality organization of the dipeptide chains regulates the formation of helical assemblies in the palladium complex composed of pyridine-dipeptide conjugate, which is found to be serve as a metalloreceptor (Figure 3.4c).
Research focused on the highly ordered structure of polypeptides has also been developed. Poly-L-lysine bearing multiple positively charged side chains (Figure 3.5a) and poly(L-glutamic acid) bearing multiple negatively charged side chains (Figure 3.5b) are demonstrated to be served as a spatially aligned polymeric scaffold for the arrangement and self-assembly of functional compounds through electrostatic interaction, resulting in the induction of specific properties. We have also succeeded in controlling the emission properties and highly ordered structure by introducing different proportions, polymer chain lengths, and different kinds of metals. Not only functional complexes, but also organic molecules can be aligned and aggregated, making it possible to control emission properties (Figure 3.5b).
Redox-Active p-Conjugated Systems for Functionalized Catalysts and Materials
The development of next-generation organic electronic materials requires molecular design based on novel concepts. The structural and electronic control of the electronic and spin states is important. For chiral luminescent materials, it is desirable to add features such as ON/OFF or switching of chiroptical properties. We also challenge the functionalization of multi-step chiroptical properties using redox states.
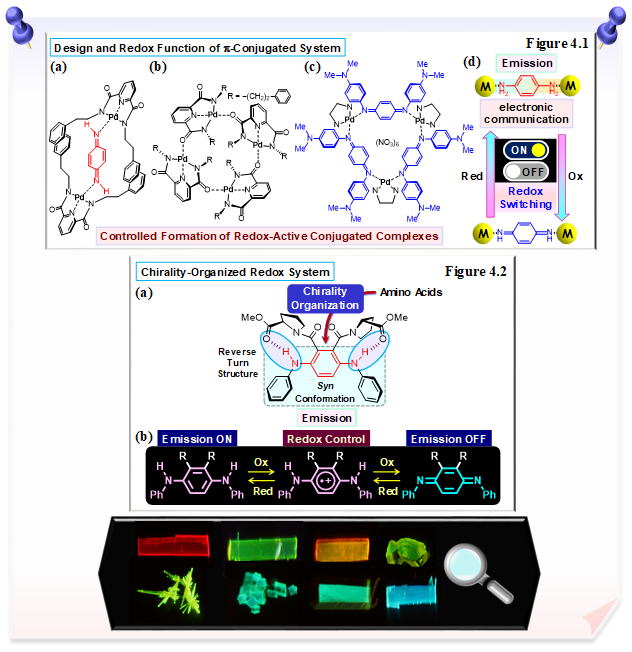
Focusing on the conductive polymer polyaniline and its constituent quinondiimine and phenylenediamine derivatives as redox-active conjugated ligands, a one-pot synthesis method for redox-active conjugated complexes is developed. Controlled formation and redox function of conjugated complexes are also possible (Figures 4.1a-d). We have also constructed the chirality-organized redox systems based on the introduction of amino acids into phenylenediamine derivatives, and have demonstrated the importance of intramolecular hydrogen bonds in stabilizing unstable radicals, chirality-organized structure and redox functionalization (Figure 4.2a). In addition, the redox state and aggregation control of the phenylenediamine moiety has been used to successfully switch the emission properties and electronic interactions (Figure 4.2b).
Contact
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University
3-3-138 Sugimoto, Sumiyoshi-ku Osaka 558-8585, Japan
Phone: +81-6-6605-2915 FAX: +81-6-6605-2500 moriuchi[at]sci.osaka-cu.ac.jp
3 min. walk from Sugimoto-cho Station (JR Hanwa Line).
15 min.walk from Abiko Station (Subway Midosuji Line).